2020 FDA Approvals: A Year in Review
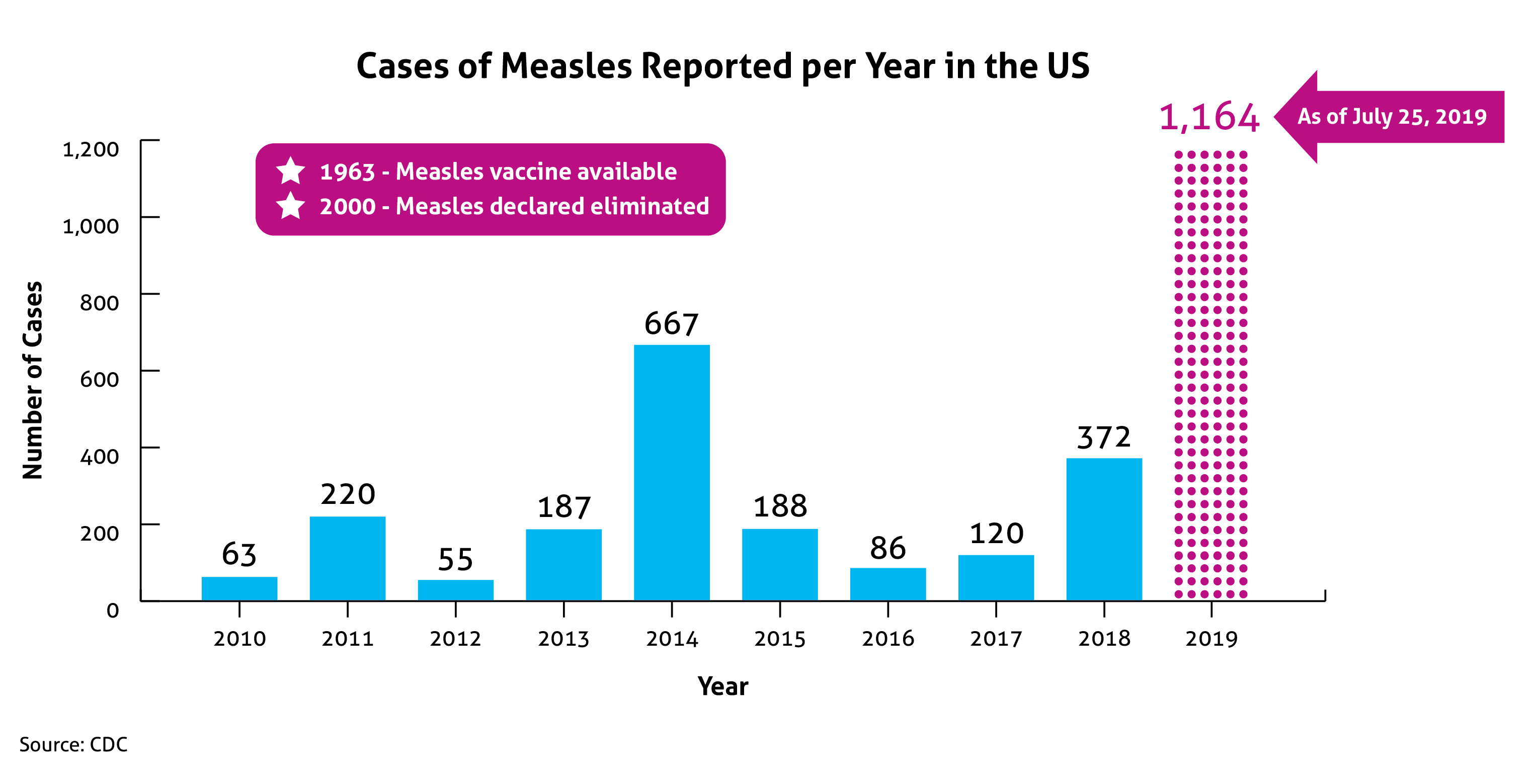
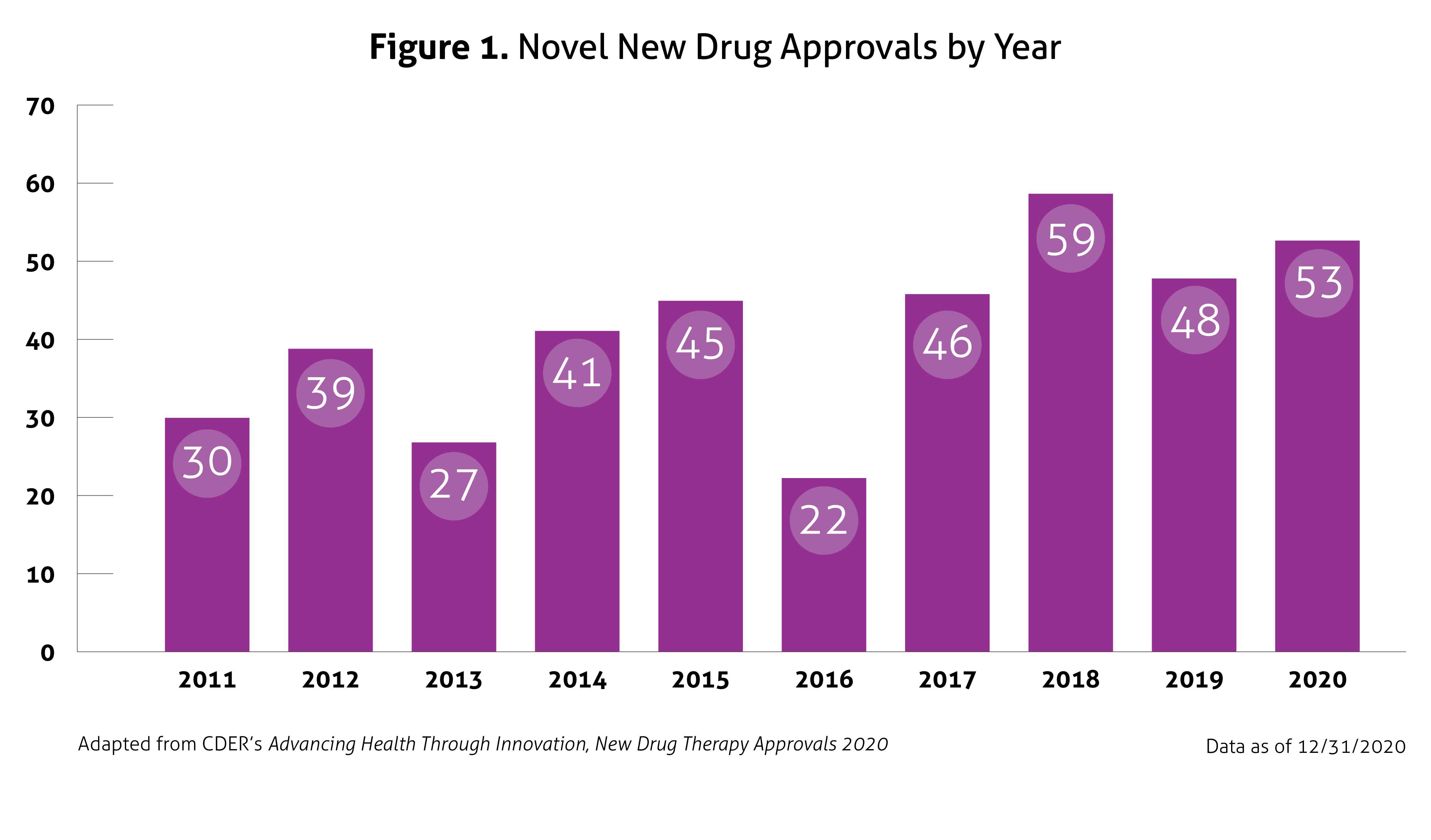
In January 2021, the United States (US) Food and Drug Administration’s (FDA) Center for Drug Evaluation and Research (CDER) published Advancing Health through Innovation: New Drug Therapy Approvals 2020. This report provides a summary of a number of approvals and highlights the novel therapies approved in 2020, continuing the generally upward trend in approval volume seen over the past decade, despite the impact of coronavirus disease 2019 (COVID-19). Compared to 2018 and 2019, in which CDER approved 59 and 48 new drugs, respectively, 53 novel agents were approved in 2020. This number does not include new and expanded uses of already approved drugs, new formulations, new dosage forms, vaccines, blood products, cellular or gene therapy, or biosimilar approvals. Once again, the number of approvals exceeded the average of 41 novel approvals per year in the past 10 years. Figure 1 outlines approvals over the past 10 years.
 Despite the ongoing pandemic the FDA continued their strategic initiatives to expedite the safe review of treatments in 2020. With the unprecedented challenges incurred in 2020, the FDA acknowledged that maintaining their commitment to bringing forth innovative therapies was difficult. Remarkably, the numbers reported by the FDA do not include the several emergency use authorizations (EUAs) issued by the FDA for COVID-19.
Despite the ongoing pandemic the FDA continued their strategic initiatives to expedite the safe review of treatments in 2020. With the unprecedented challenges incurred in 2020, the FDA acknowledged that maintaining their commitment to bringing forth innovative therapies was difficult. Remarkably, the numbers reported by the FDA do not include the several emergency use authorizations (EUAs) issued by the FDA for COVID-19.
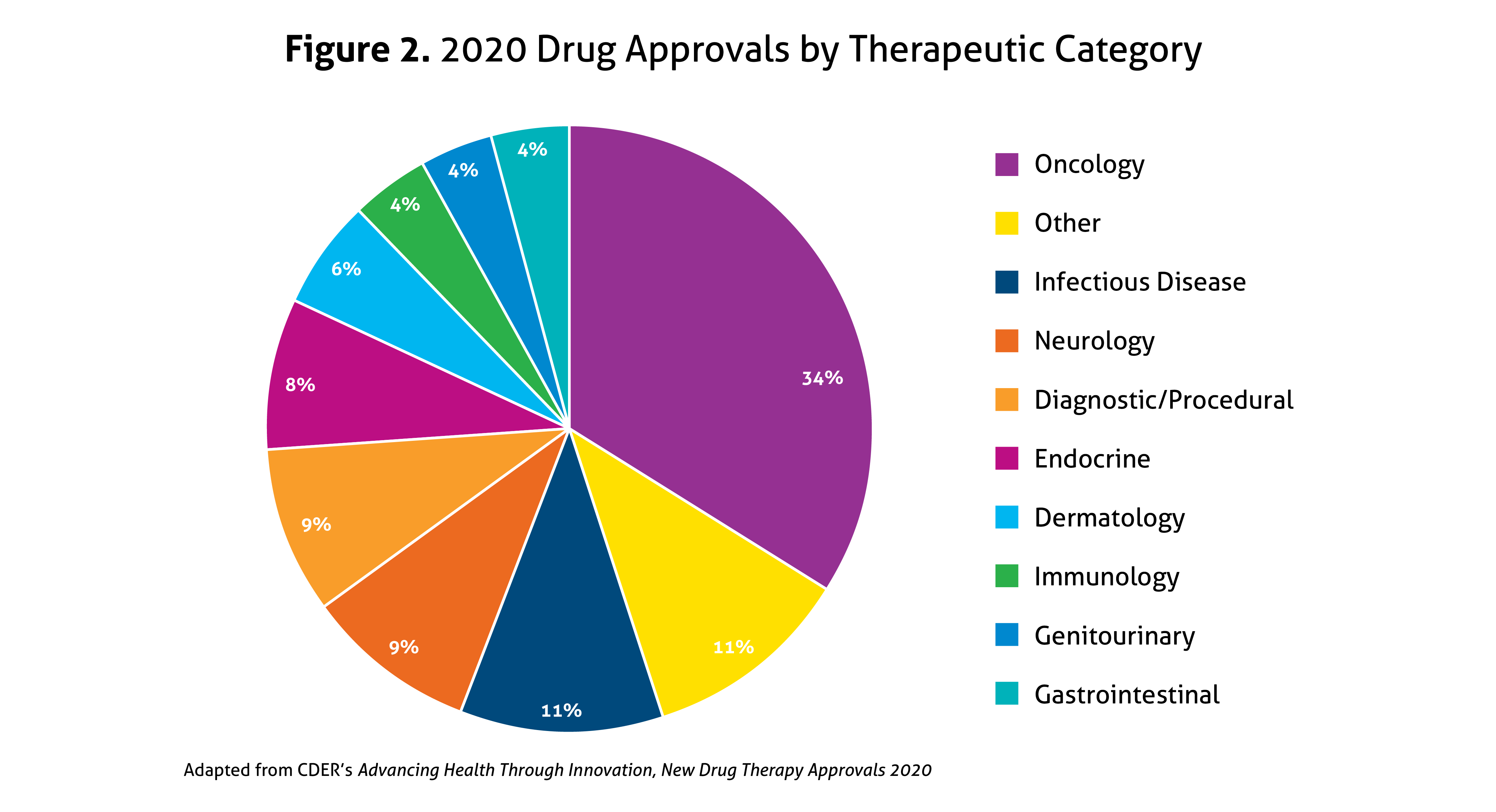
Last year, all 53 novel drug approvals again met their Prescription Drug User Fee Act (PDUFA) goal dates, cementing this as a priority for the Agency. In 2020, 40% were considered first-in-class, and 58% were approved for rare diseases (Orphan Drugs), the latter of which increased from 44% in 2019. Priority Review was granted to 57% of novel drugs, 23% received Accelerated Approval, 42% were designated as Breakthrough Therapy (up from 27% in 2019), and 32% garnered Fast Track designation. Overall, 68% of all drug approvals in 2020 used expedited development and review methods. In addition, 92% were approved in the first review cycle, and 75% were approved in the US prior to receiving approval in other countries. A breakdown of the types of drugs approved in 2020 is illustrated in Figure 2, with agents within the oncology spectrum representing over one-third of 2020’s novel approvals.
The notable 2020 approvals encompassed new advances for the treatment of infectious diseases, including a new medication class for the treatment of human immunodeficiency virus-1 (HIV-1). Garnering perhaps the most attention, the FDA also approved the first medication for hospitalized patients with COVID-19. Unique infectious diseases in the US also received attention, with a new drug for malaria, two new options for the Ebola virus, and a new treatment for Chagas disease approved in 2020. In the neurology arena, there were multiple approvals of agents for more common conditions, such as migraine or Parkinson’s disease. Moreover, there were significant advances for rare neurological conditions, including the first oral agent for spinal muscular atrophy and new treatments for rare seizure disorders. In addition, two immunological agents were approved for the treatment of neuromyelitis optica spectrum disorder. Additional treatment options emerged for several autoimmune conditions in 2020 as well. Not surprisingly, numerous advances were made within the oncology umbrella, including both novel approvals and new or expanded indications for several existing agents. With over half of the novel approvals being classified as Orphan Drugs, in 2020, the FDA has fortified their dedication to providing innovative and often targeted treatment options for all individuals.