The path to appropriate biosimilar management
Magellan Rx has been a market leader in developing forward-thinking solutions to combat rising specialty spend on the medical benefit for nearly 20 years. With a passion for solving complex pharmacy challenges, such as biosimilar management, we roll up our sleeves and tackle what is truly driving trend while ensuring a high quality of care for the members we serve.
Building on the success of our industry-first medical pharmacy program, including management of medical benefit oncology drug spend, we began to focus on advancing biosimilar utilization in 2015. Our goal was to empower health plan customers with education and strategies that turned biosimilar availability into cost savings while maintaining clinical quality. From the high-cost autoimmune category to oncology and beyond, our philosophy to biosimilar management involves three key components:
- Proactive Management: Assessing and developing clinical protocols while educating and communicating with network providers
- Medical Pharmacy Execution: Leveraging Magellan Rx’s innovative medical management expertise by incorporating biosimilars into key utilization management programs such as medical prior authorization and provider reimbursement/fee schedule management
- Expert Opinion: Continuously working to gain insights from our advisory board of specialists and Expert Clinical Network of key opinion leaders
But first, what is a biosimilar?
According to the U.S. Food and Drug Administration (FDA), a biosimilar is a drug type that is highly similar to an FDA-approved biologic, or reference product, with regards to its purity, molecular structure, and bioactivity. The biosimilar approval pathway starts with an application submission that includes analytical studies, animal studies, and at least one clinical study. A biosimilar is approved by the FDA after evaluation and testing to show it is as safe and effective as its reference product.
As of April 2021, there are now 29 FDA-approved biosimilar products across three different categories—20 have been launched to date, and 18 are oncology or oncology support.

Magellan Rx’s approach
In 2015, ahead of the first biosimilar approval in the U.S., our work began with a committee of experts to review the biosimilar landscape and potential impacts for payers. By 2016, we had established our first biosimilar-over-reference policy on the medical benefit, and in 2017 additional clients opted into the strategy with more growth in biosimilar savings.
The program was expanded in 2018 to include infliximab—the biosimilar for Remicade, a top spend drug used to treat autoimmune conditions such as rheumatoid arthritis and inflammatory bowel disease—by leveraging a comprehensive utilization management solution. As part of this initiative, our team of highly-trained pharmacists worked with physician offices and hospitals to ensure appropriate utilization for each patient’s unique situation.
Also in 2018, we established an Oncology Biosimilar Workgroup to prepare for future launches in this high-spend category (in fact, oncology and oncology support accounts for more than 40% of total medical pharmacy spend across the Commercial, Medicare, and Medicaid lines of business1). We aimed to educate health plan customers, members, and providers through individualized strategies that consider clinical, financial, and regulatory factors. The oncology biosimilar program was launched in 2019 as oncology biosimilars hit the market, with early adopter implementation that resulted in maintaining or expanding member access to clinically-effective treatments while delivering significant drug spend savings.

Due to the success of the infliximab program and proactive approach to the launch of oncology biosimilars, we experienced a rapid expansion in 2020 as clients, representing millions of lives, began to adopt these innovative solutions. Oncology biosimilar utilization surged, and the (measured by the number of prior authorizations approved for the first two therapies with biosimilars compared to the reference brands) for early adopters. We also showcased results in research presented at the 2020 AMCP Annual and AMCP NEXUS industry events.
So far in 2021, biosimilar uptake continues to increase. Our team of experts previewed results from the oncology biosimilar program at the 2021 AMCP Annual event and spoke to The Center for Biosimilars on strategies that payers are using to promote biosimilar adoption.
Looking ahead, Magellan Rx remains committed to the biosimilar-first strategy and will continue to expand the program to include additional categories and available biosimilar agents as they are approved by the FDA. For more on payer management concerns related to biosimilars, read page 24 of the latest Magellan Rx Management Trend Report. For up-to-date pipeline news, check out the MRx Pipeline Report. Have questions or want to implement a solution to meet the needs of your unique population? Connect with us today!
To learn more about Magellan Rx’s work, click here.
- Magellan Rx Management Medical Pharmacy Trend Report™, © 2021.
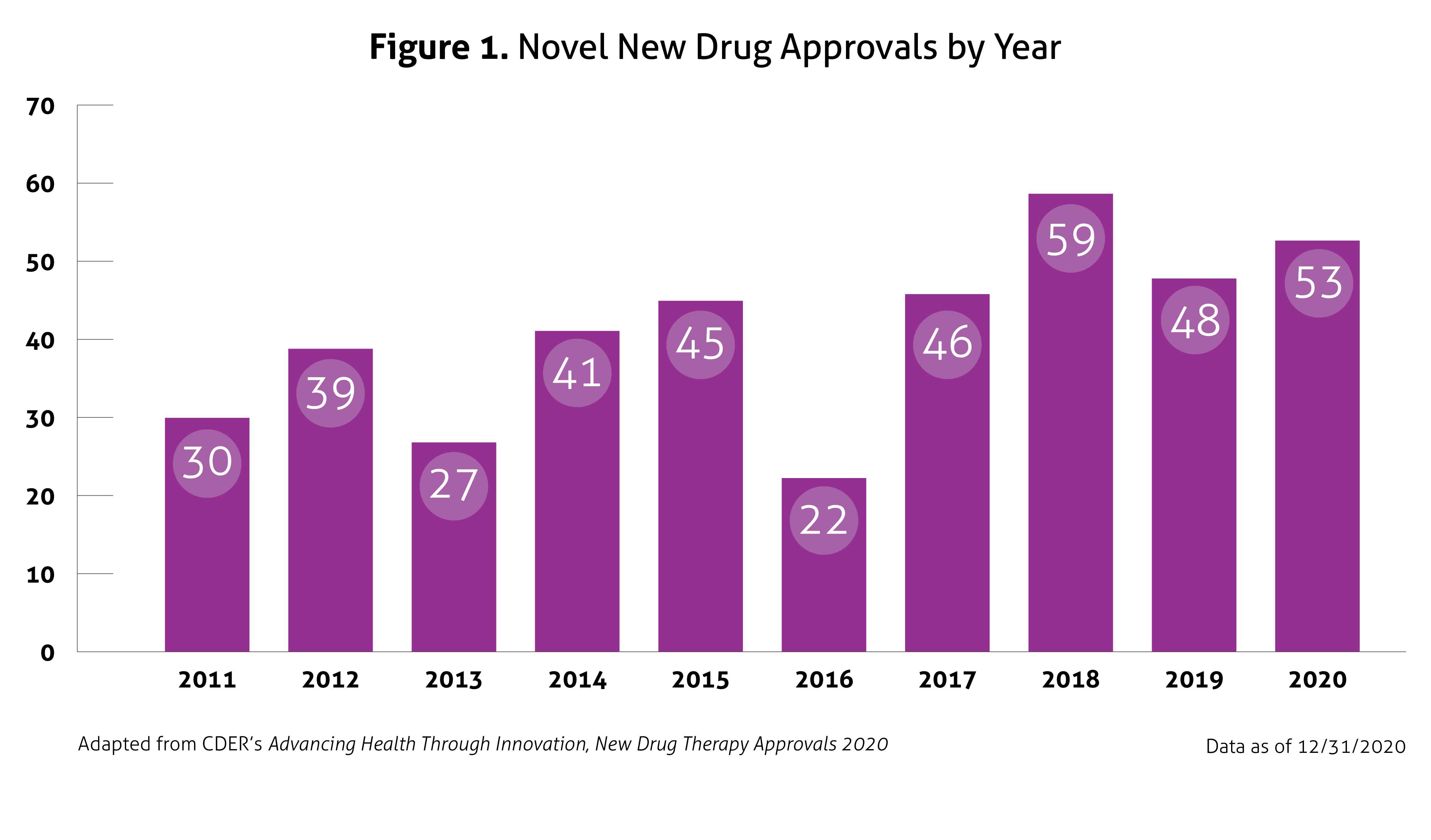 Despite the ongoing pandemic the FDA continued their strategic initiatives to expedite the safe review of treatments in 2020. With the unprecedented challenges incurred in 2020, the FDA acknowledged that maintaining their commitment to bringing forth innovative therapies was difficult. Remarkably, the numbers reported by the FDA do not include the several emergency use authorizations (EUAs) issued by the FDA for COVID-19.
Despite the ongoing pandemic the FDA continued their strategic initiatives to expedite the safe review of treatments in 2020. With the unprecedented challenges incurred in 2020, the FDA acknowledged that maintaining their commitment to bringing forth innovative therapies was difficult. Remarkably, the numbers reported by the FDA do not include the several emergency use authorizations (EUAs) issued by the FDA for COVID-19.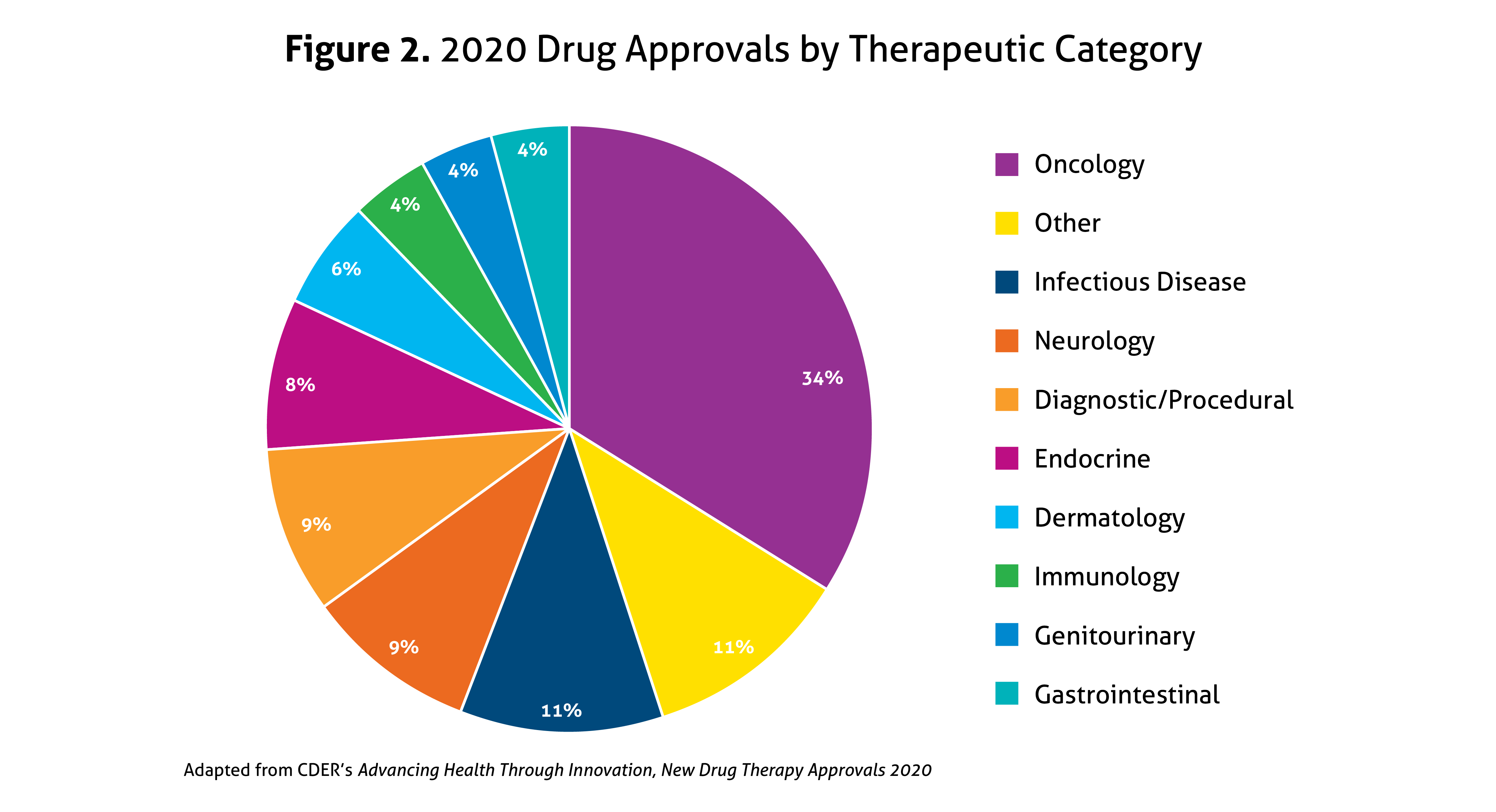
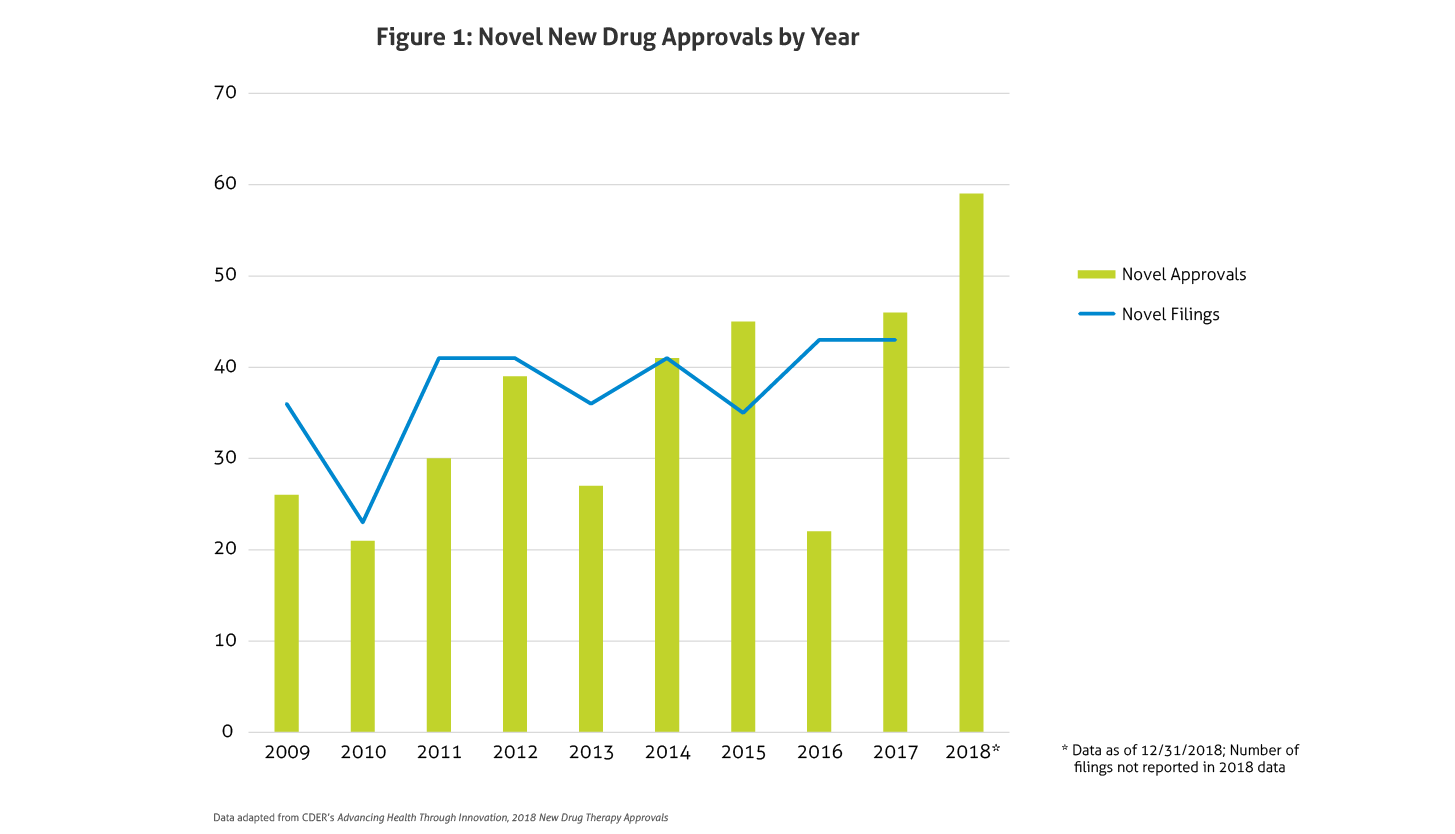
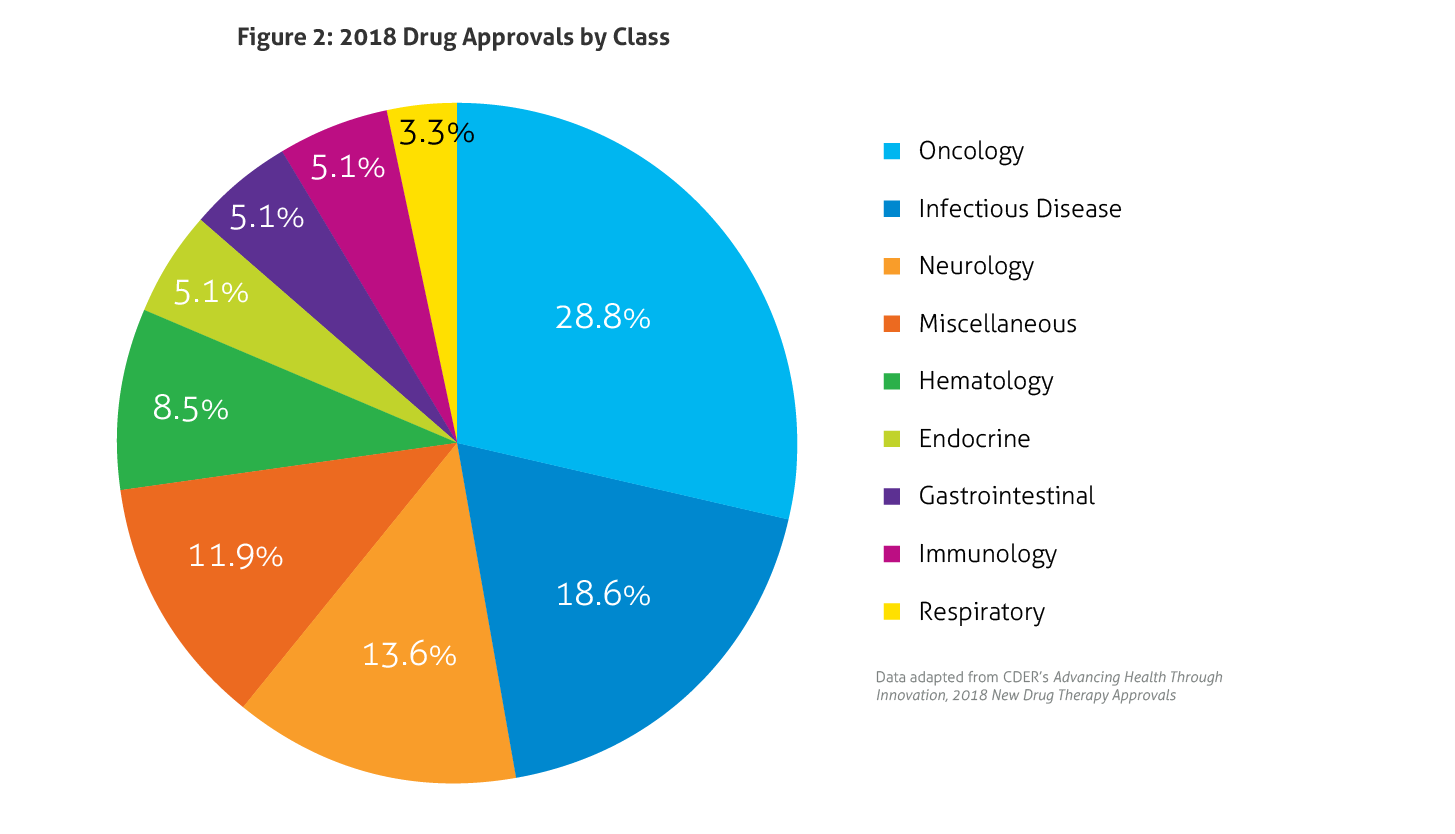 Some of the notable 2018 approvals included the first non-opioid drug approved to reduce opioid withdrawal symptoms, a new antiretroviral for multidrug resistant human immunodeficiency virus-1, a new class of drugs for migraine (calcitonin gene-related peptide receptor antagonists), the first FDA-approved drug derived from marijuana, the first treatment approved for multiple sclerosis in children, expanded options for cystic fibrosis, and the first antibiotic approved under the Limited Population Pathway for Antibacterial and Antifungal Drugs.
Some of the notable 2018 approvals included the first non-opioid drug approved to reduce opioid withdrawal symptoms, a new antiretroviral for multidrug resistant human immunodeficiency virus-1, a new class of drugs for migraine (calcitonin gene-related peptide receptor antagonists), the first FDA-approved drug derived from marijuana, the first treatment approved for multiple sclerosis in children, expanded options for cystic fibrosis, and the first antibiotic approved under the Limited Population Pathway for Antibacterial and Antifungal Drugs.